fastq文件怎么打开 fastaq,fq文件如何打开教程
更新时间:2023-06-05 16:08:32作者:yang
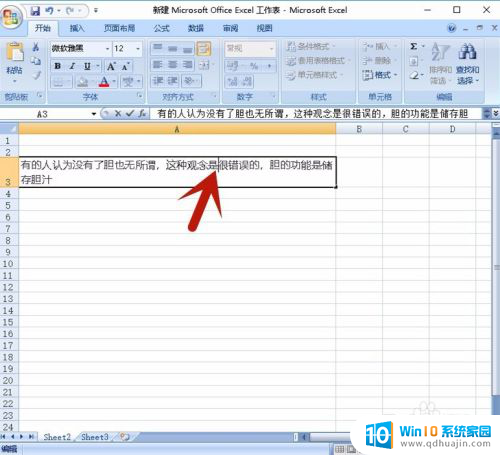
现在科研中常用的高通量测序技术下机后会生成大量的fastq格式文件,而要分析这些数据就必须要打开这些文件。那么fastq文件怎么打开呢?常见的打开方式是使用文本编辑器,但这样看起来不直观、容易混淆。今天我们就来讲解一下fastq或fq格式文件如何打开,以便更好地处理后续数据。
fastaq,fq文件如何打开教程
生命科学研究生的苦逼在于:一切从零开始!什么是fastaq结尾的文件?fastaq结尾的文件是Illmina公司二代测序结果输出的标准文件,其中每一条序列信息包括4行:
@HWUSI-EAS100R:6:73:941:1973#0/1 GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGT +HWUSI-EAS100R:6:73:941:1973#0/1 !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC6
简单来讲,@开头的第一列是reads的ID以及其他信息。第二列是序列信息。+开头的第三列是跟随该read的名称(一般于@后面的内容相同),但有时可以省略,但“+”一定不能省。第四行代表reads的质量
如何打开fq文件呢?有多种方式可以打开fq文件: 1. 最为原始的是在Linux服务器上用查看文本的less命令或cat命令例如:
less [参数]... 目标文件 参数: -S:单行显示 -N:行号加入编号 示例:less -S -N ~/.bashrc 实例: zless -S SRR1039510_1.fastq.gz #由于是压缩文件用zless打开压缩的fq文件,-S一定大写,注意!空格键表示继续查看,按q键表示退出 #结果如下 @SRR1039510.1 HWI-ST177:290:C0TECACXX:1:1101:1 TGGGAGGCTGAGGCAGGAGAATCACTTAAACCTGGGAGGCAGAGGT +SRR1039510.1 HWI-ST177:290:C0TECACXX:1:1101:1 HJJJIJJJJJJJJIJJJGHHIJIIIIIIJJEHGGIJGIJIJJIJHH @SRR1039510.2 HWI-ST177:290:C0TECACXX:1:1101:1 AAAGAAGGCGACAGTGAGAAGGAGTCCGAGAAGAGTGATGGAGACC +SRR1039510.2 HWI-ST177:290:C0TECACXX:1:1101:1 HJJJJJJJJJJJIJIIGIJJJJGJHJJJHHDFFFE@CEEEDDDDDD cat [参数] 文本... # 注意cat一次打开全部文本,如果文件太大会导致服务器卡死,需要联合head命令和tail命令使用,打开文件的头部和尾部 参数: -n: 按行数编号 -A:等价于-vET 实例: zcat SRR1039510_1.fastq.gz|head -n 10 # |是管道符用于传递数据,head用于查看文件的头部数据,可以定义查看头几行 #结果 @SRR1039510.1 HWI-ST177:290:C0TECACXX:1:1101:1373:2104 length=63 TGGGAGGCTGAGGCAGGAGAATCACTTAAACCTGGGAGGCAGAGGTTACAGTGAGCCGAGATT +SRR1039510.1 HWI-ST177:290:C0TECACXX:1:1101:1373:2104 length=63 HJJJIJJJJJJJJIJJJGHHIJIIIIIIJJEHGGIJGIJIJJIJHHHGGFFDFFFDEDDDBDC @SRR1039510.2 HWI-ST177:290:C0TECACXX:1:1101:1340:2124 length=63 AAAGAAGGCGACAGTGAGAAGGAGTCCGAGAAGAGTGATGGAGACCCAATAGTCGATCCTGAG +SRR1039510.2 HWI-ST177:290:C0TECACXX:1:1101:1340:2124 length=63 HJJJJJJJJJJJIJIIGIJJJJGJHJJJHHDFFFE@CEEEDDDDDDDDDDDDDDDBDDDDDDD @SRR1039510.3 HWI-ST177:290:C0TECACXX:1:1101:1273:2183 length=63 CTGCTGGGCCCCAAGGTCCTCCTGGTCCCAGTGGTGAAGAAGGAAAGAGAGGCCCTAATGGGG同时也可以用pycharm软件打开fq文件,直接拖入打开即可 ###现在我们知道fq的数据是如何打开了,然而我们通常进行处理的文件是fasta文件,如何将fq无损的转化成fasta文件? 有以下几种方式可以将fq格式文件转化成fasta文件:在Linux服务器上用基础命令来实现这个转换:
zless -S SRR1039510_1.fastq.gz| paste - - - -|cut -f 1-2|tr '\t' '\n'|tr '@' '>' |less -S > SRR1039510_1.fasta #结果为: >SRR1039510.1 HWI-ST177:290:C0TECACXX:1:1101:1373:2104 length=63 TGGGAGGCTGAGGCAGGAGAATCACTTAAACCTGGGAGGCAGAGGTTACAGTGAGCCGAGATT >SRR1039510.2 HWI-ST177:290:C0TECACXX:1:1101:1340:2124 length=63 AAAGAAGGCGACAGTGAGAAGGAGTCCGAGAAGAGTGATGGAGACCCAATAGTCGATCCTGAG >SRR1039510.3 HWI-ST177:290:C0TECACXX:1:1101:1273:2183 length=63 CTGCTGGGCCCCAAGGTCCTCCTGGTCCCAGTGGTGAAGAAGGAAAGAGAGGCCCTAATGGGG >SRR1039510.4 HWI-ST177:290:C0TECACXX:1:1101:1562:2147 length=63 CTTGGCTGCAGCCATCCCGCTTAGCCTGCCTCACCCACACCCGTGTGGTACCTTCAGCCCTGG # 同时生成SRR1039510_1.fasta文件。 # 同时zcat SRR1039510_1.fastq.gz| paste - - - -|cut -f 1-2|tr '\t' '\n'|tr '@' '>' |less -S 也可实现这种转换如果你不需要查看fq中的具体内容,可以用软件实现fq到fasta文件的转换。fastX_tool软件可以实现相同的功能
fastq_to_fasta -Q33 -v -n -i SRR1039510_1.fastq -o SRR1039510_1.fasta #其中-Q33是必须添加的参数
如遇生信方面的技术困难欢迎咨询猫叔!

总的来说,打开fastq或fq文件很简单。只需要使用适当的文本编辑器或者基因组学软件即可。无论是查看序列数据还是进行序列分析,打开fastq/fq文件都是科学研究中重要的一步。希望本文中提供的教程能够帮助到需要的读者。