gemtc软件下载 生存数据的贝叶斯网状Meta分析R包gemtc使用方法
随着临床研究的不断进步,越来越多的研究结果涌现,而这些结果往往主要包括生存数据。要真正从这些数据中提取出有用的信息,则需要使用一些数据分析工具。而在这方面,Meta分析以其综合多项研究的结果并增强数据可靠性的特点,成为了热门分析方法之一。而针对生存数据的Meta分析,则需要专门的工具,在这里介绍的gemtc软件就是其中之一。本文将围绕gemtc软件下载以及其使用方法展开探讨,帮助读者了解更多关于这一工具的信息。
生存数据的贝叶斯网状Meta分析R包gemtc使用方法
前提条件:准备工作完成后,打开RStudio后,在console里输入install.packages(“gemtc”)
第一次安装的时候,安装速度可能比较慢,耐心等待一下。
等安装完成后,设置我们的工作目录。后面可以把待分析的数据放入这个工作目录,敲入:
setwd("D:\\R网状教程\\HR") #你可以更改为自己的工作目录,在后面环境变量增多的时候,在这一步前可以加一个rm(list=ls())清除环境变量。
然后敲入:
library(gemtc) #装载gemtc包
关于如何将生存数据准备成可以用R的GEMTC分析的格式,以及如何计算多臂研究中参考臂的标准误,请参考公众号内教程。
正式分析把刚刚准备好的数据导入到R里,敲入:
data <- read.csv("sample1.csv", sep=",", header=T) #sep代表逗号为分隔符,header=T代表把首行的数据作为标题,<-是R里的赋值符号,你可以把它理解为=号。<-前面的变量名是可以自己任意指定的,以下类似。把数据弄成gemtc的网络格式,敲入:
network <- mtc.network(data.re=data) #这里<-前面的network不一定非要叫network,改为其他的阿猫阿狗名称均可。以下类似,不再赘述。
输入:plot(network)
可以看到网状图如下:

当然了,这些大小、颜色、字体等等的参数都是可以调整的,但是跟stata做网状图相比还是太麻烦了。Stata几乎不用调整一步到位,大家可以参考我之前制作的如何使用stata对生存数据做网状关系图的教程。
然后开始建立模型:
model <-mtc.model(network, type="consistency", n.chain=4,likelihood="binom",link="cloglog",linearModel="random")
其中,圆括号里的第一个参数为我们刚刚新建的gemtc网络类型的变量network,第二个参数type="consistency",是指我们的模型为一致性模型,n.chain指的是马尔科夫蒙特卡洛MCMC的链条数目,一般为2-4均可。因为我们进行的是生存数据的网状分析,所以likelihood="binom", link="cloglog"。linearModel="random"指的是我们使用的模型为随机效应,当然也可以指定fixed作为固定效应模型。这一步可能会跳出一些红色的warning,强迫症的同学看到可能会感到不安,但不用紧张,不影响后续的分析,不管它即可。
现在就可以进行迭代了,敲入:
results <- mtc.run(model, n.adapt = 20000, n.iter = 50000, thin = 1)
其中,model是我们上一步建立的模型名称,n.adapt为退火次数,n.iter为迭代次数。这个时候我们耐心等待,RStudio界面如下所示:
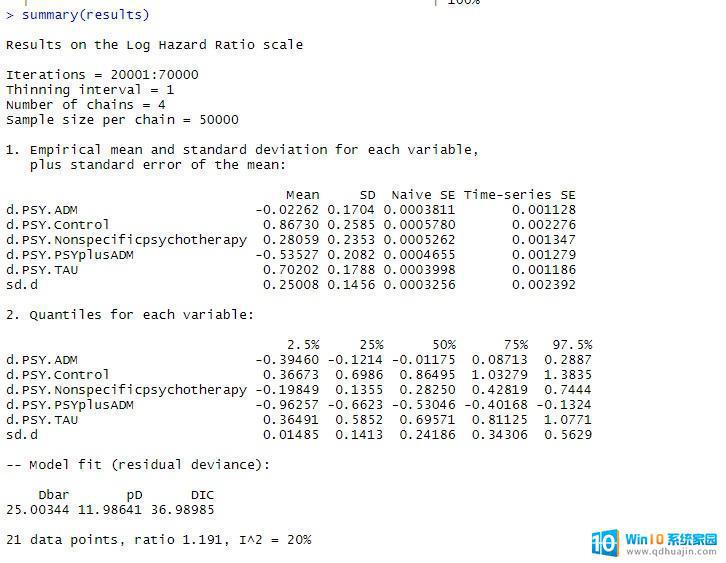
敲入:summary(results)可以看到如下结果:
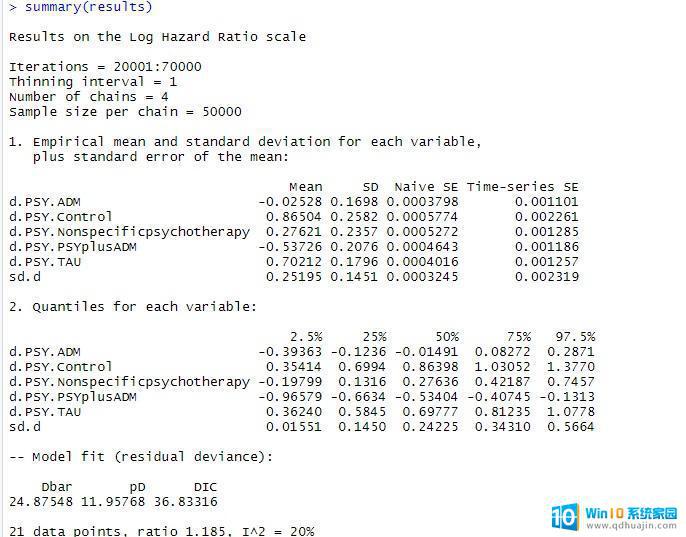
其中结果包括DIC,DIC可以进一步与不一致性模型的DIC作比较,如果相差在5以内,就说明数据基本符合一致性的前提。DIC的绝对值没有任何意义,只用于相对比较。以及I2这个总体异质性参数。总体异质性参数也可以报告tau2,上图中的sd.d就是tau,本例中为0.25,那么tau2就是0.06。
敲入:
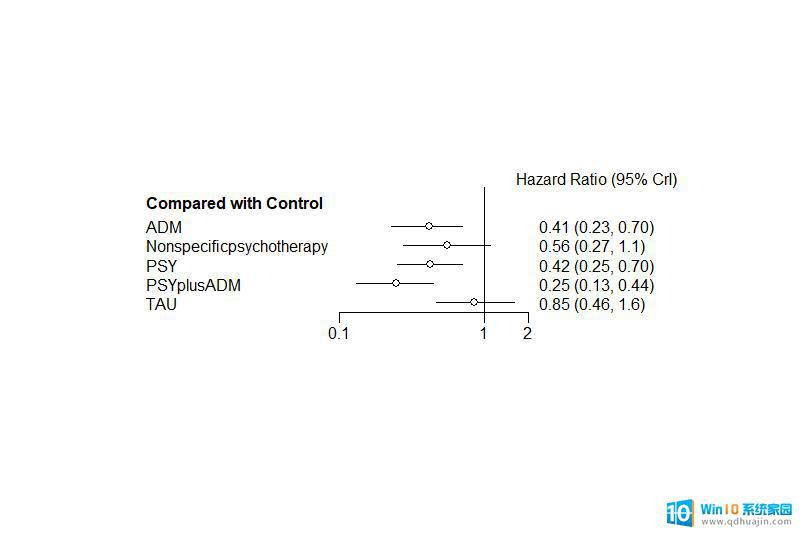
forest(relative.effect(results, "Control"))
可以看到每个干预相对于Control的森林图:
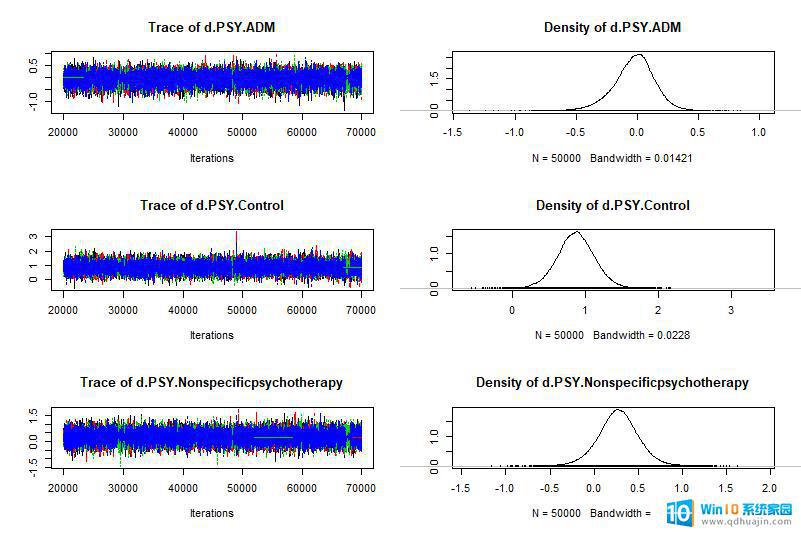
敲入:plot(results) #可以看到每个指标的收敛情况以及密度图
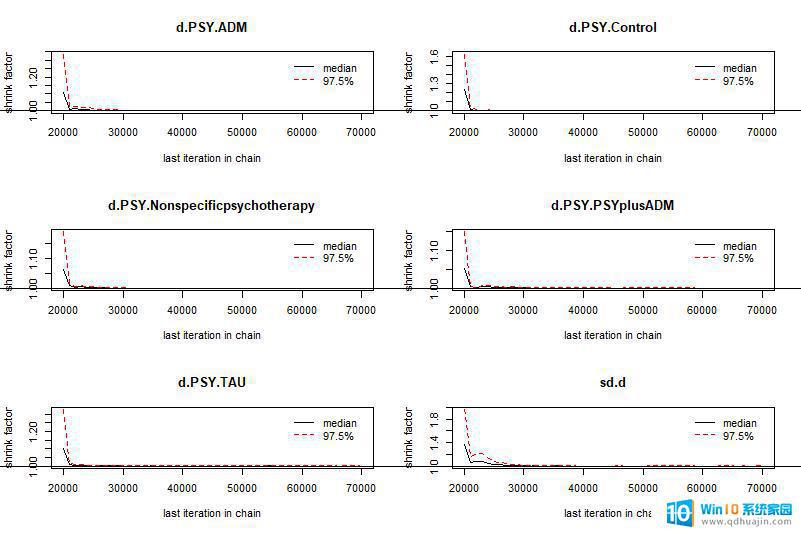
敲入:gelman.plot(results) # 收敛性诊断
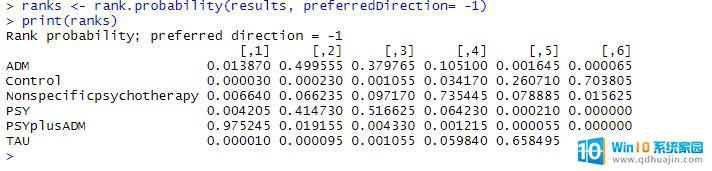
对所有的干预措施进行排序,敲入:
ranks <- rank.probability(results, preferredDirection= -1) #这里的效应量越小越好,所以direction选择-1,默认为效应量越大越好 print(ranks)
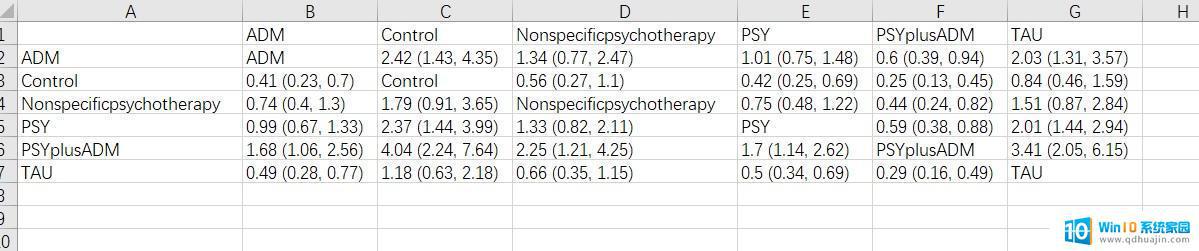
制作联赛表:
a <- round(exp(relative.effect.table(results)),2) write.csv(a, "leaguetable.csv")
以上步骤可以在工作目录中导出这个表格。
 全局不一致(Global inconsistency)
全局不一致(Global inconsistency)很多同学不知道如何做不一致模型,想当然的以为将type="inconsistency"就OK了,其实并非如此。而应是在模型那一步中将type="ume"或者"use"。至于ume和use具体是什么,本文不再赘述,感兴趣的同学可以查看交流群文件中TSD4(inconsistency)的文件。如下:
modelume <-mtc.model(network, type="ume", n.chain=4, likelihood="binom", link="cloglog", linearModel="random") resultsume <- mtc.run(modelume, n.adapt = 20000, n.iter = 50000, thin = 1) summary(resultsume) #查看不一致模型的DIC,然后与一致性模型进行比较。
 局部不一致(local inconsistency)
局部不一致(local inconsistency)用节点劈裂法
resultnodesplit <-mtc.nodesplit(network, n.adapt = 20000, n.iter = 50000, thin = 1, n.chain=4, likelihood="binom", link="cloglog",linearModel="random")
在这里,我们无需像在WINBUGS或者OpenBUGS软件里对初始值进行繁琐的设定。这一点是我非常喜欢R软件gemtc包的地方。
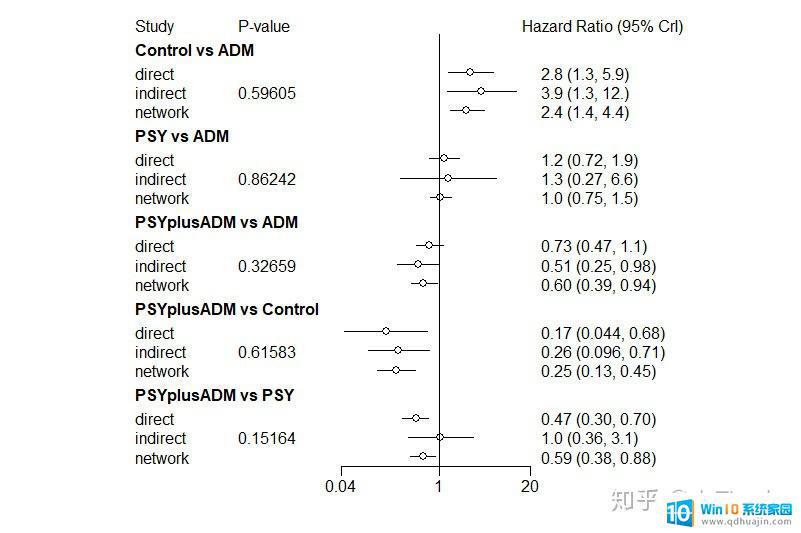
b<-summary(resultnodesplit) #查看节点劈裂法的结果 print(b)

plot(b)
 异质性分析
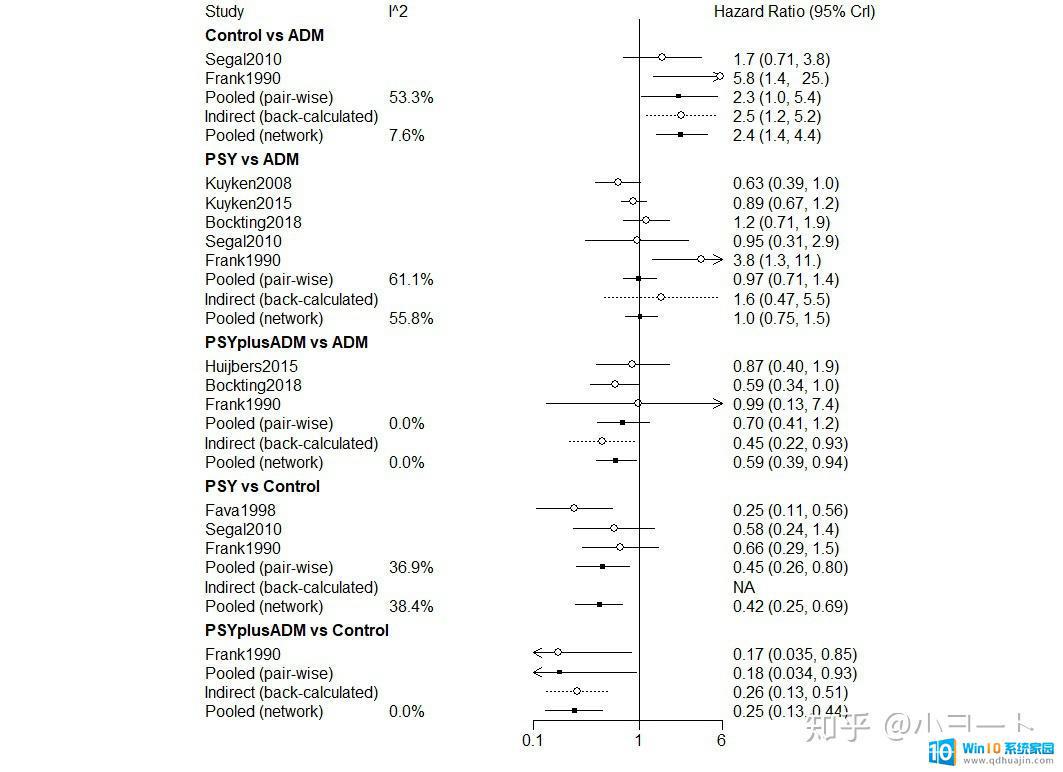
异质性分析resultanohe <- mtc.anohe(network, n.adapt = 20000, n.iter = 50000, thin = 1, n.chain=4, likelihood="binom", link="cloglog",linearModel="random") c<-summary(resultanohe) plot(c)
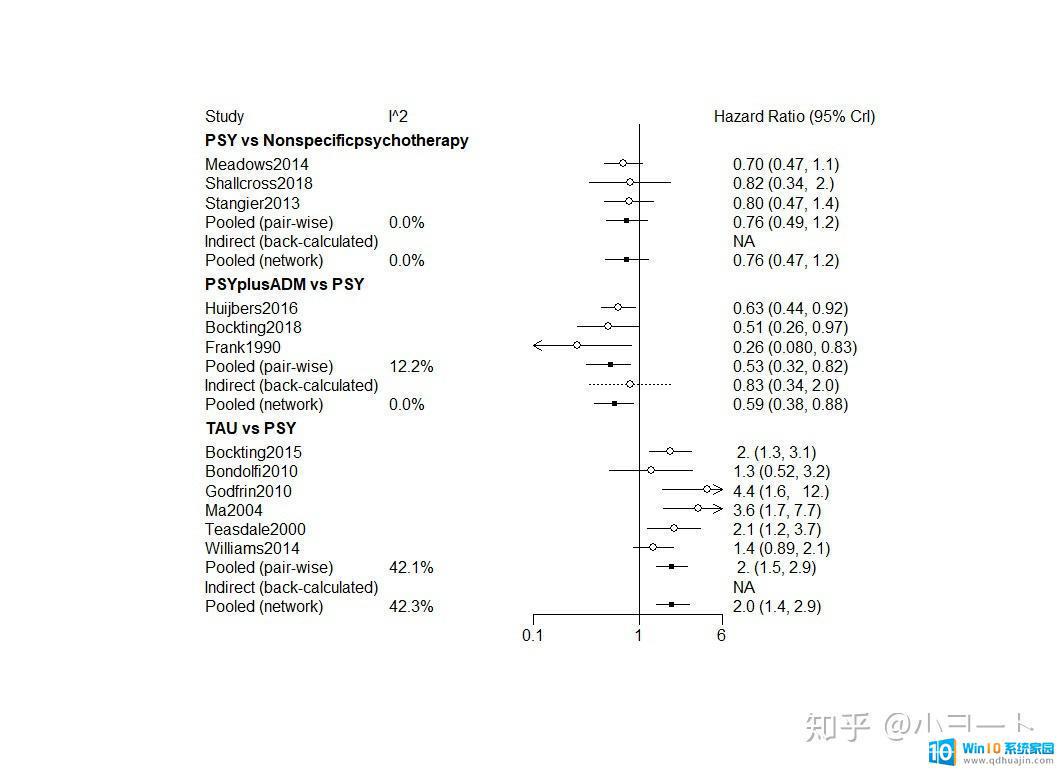
至此,主分析的步骤基本已经完成。此外,gemtc还支持对协变量进行回归分析,对生存数据的回归分析操作类似于既往的回归分析教程。
更多精彩,敬请关注
使用gemtc软件进行生存数据的贝叶斯网状Meta分析可以更全面地评估不同研究的结果,并提高研究的可靠性和重现性。通过了解gemtc的使用方法,我们可以更好地应用这一工具来帮助我们的研究得出更准确的结论。